KOBAS on CyVerse¶
Accessing KOBAS in the Discovery Environment¶
- Create an account on CyVerse (free). The Discovery Environment (DE) the user guide can be found here.
- Open the CyVerse Discovery Environment (DE) and login with your CyVerse credentials.
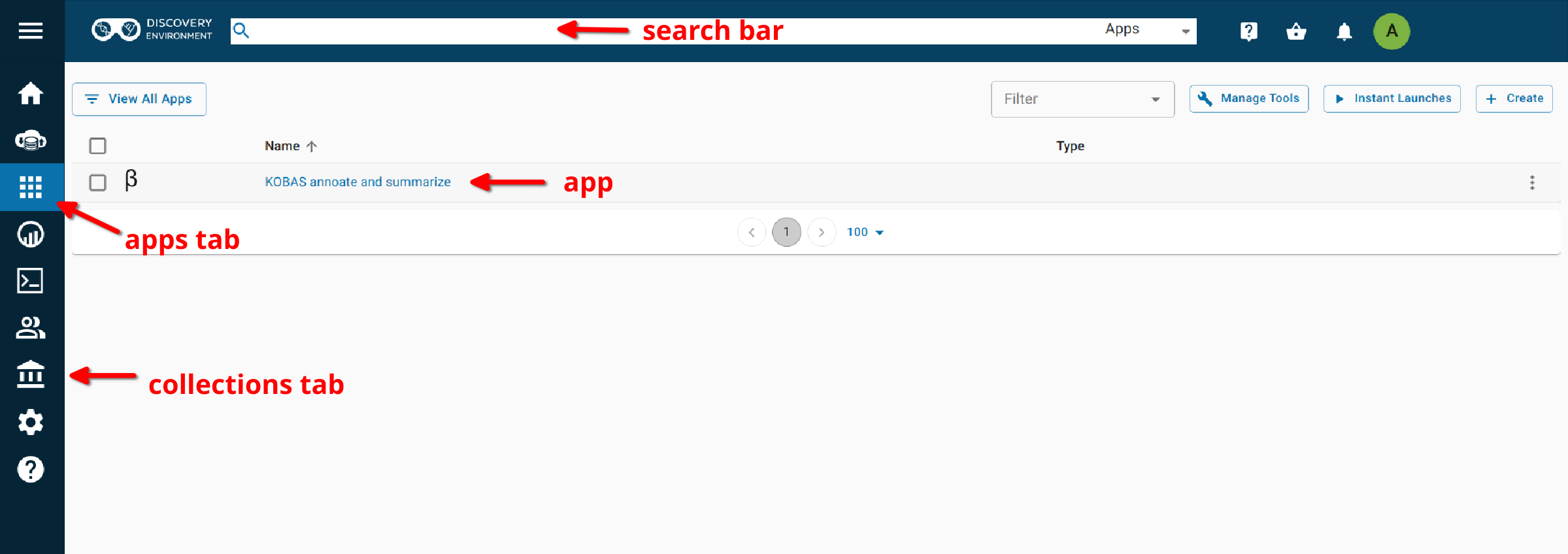
- There are several ways to access the GOanna app:
- Use the direct link.
- Search for ‘KOBAS in the search bar at the top of the ‘apps’ tab.
- Follow the AgBase collection (collections tab on left side of DE)
The KOBAS apps are called:
- NEW KOBAS annotate and summarize
- KOBAS annotate 3.0.3
- KOBAS identify 3.0.3
- KOBAS annotate and identify 3.0.3
NEW KOBAS annotate and summarize
The new KOBAS annotate and summarize app is version 3.0.3. In addition to the annotate function it also performs a summary step. We recommend using this app rather than the annotate 3.0.3 app.
KOBAS annotate and summarize¶
Launching the app¶
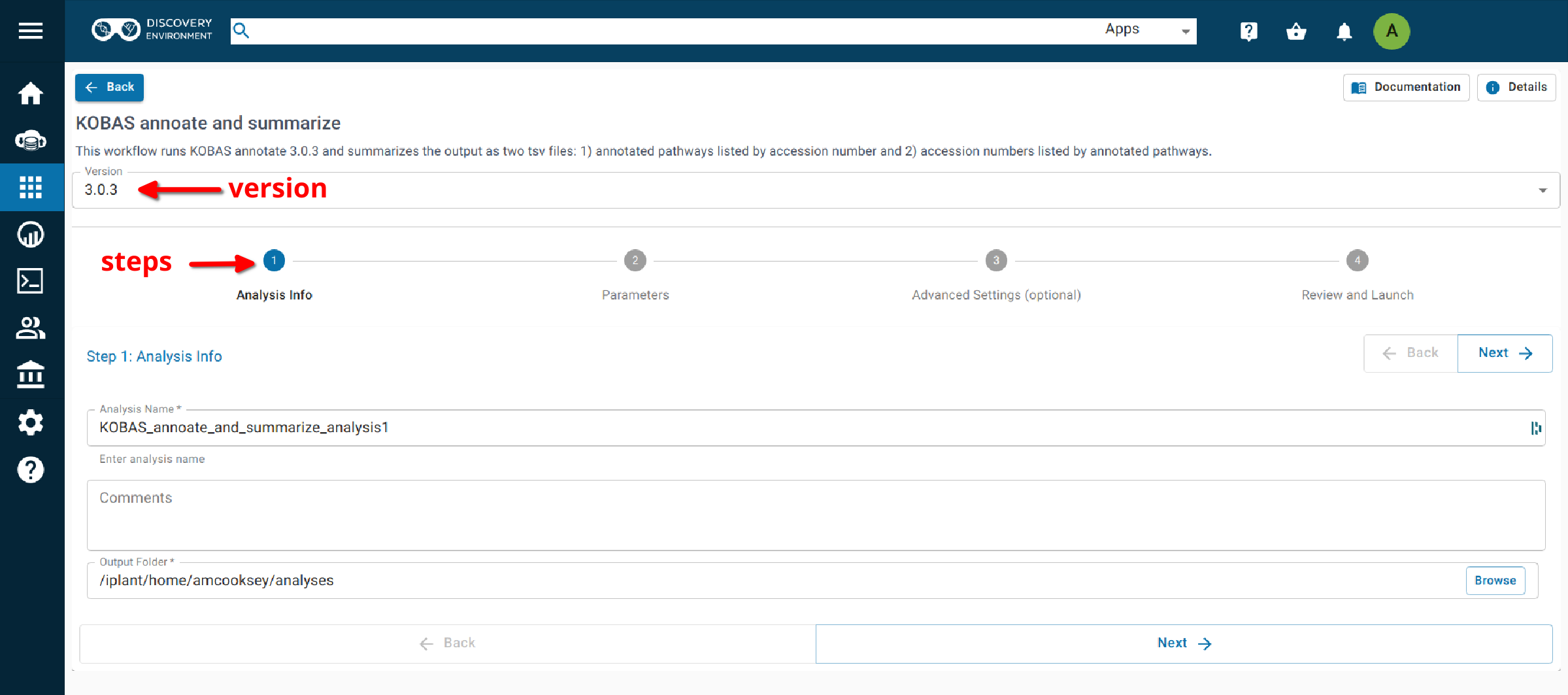
Step 1. Analysis Info¶
Analysis Name: KOBAS_annoate_and_summarize_analysis1: This menu is used to name the job you will run so that you can find it later. Analysis Name: The default name is “KOBAS_annoate_and_summarize_analysis1”. We recommend changing the ‘analysis1’ portion of this to reflect the data you are running.
Comments: (Optional) You can add additional information in the comments section to distinguish your analyses further.
Select output folder: This is where your results will be placed. The default (recommended) is your ‘analyses’ folder.
Step 2. Parameters¶
Input File: Use the ‘browse’ button on the right side of the field to navigate to your input file.
Input File Type: Select your input file type from the drop-down list. If your file type isn’t there then the app does not support that file type.
Species Code: Enter the species for the species of the sequences in your input file.
Note
If you don’t know the code for your species it can be found here: https://www.kegg.jp/kegg/catalog/org_list.html Not all KEGG species are available through KOBAS.
If your species of interest is not available then you should choose the code for the closest-related species available
E value: This is the evalue to use in the BLAST search. The default is 1e-5.
Rank: rank cutoff for valid hits from BLAST result. Default is 5.
Covergage: subject coverage cutoff for BLAST. Default is 0.
Ortholog: when checked KOBAS will only use orthologs for cross species annotation.
Output File Name: Provide an output file name .
For information on outputs see Understanding Your Results: Annotate
Step3. Adavanced Settings (optional)¶
This page allows you specifiy compute requirements for your analysis (e.g. more memory if your analysis is particularly large). You should be able to leave the defaults for most analyses.
Step4. Review and Launch¶
This will display all of the parameters you have set (other than default). Missing information that is required will displayed in red. Make sure you are happy with your choices and then clicke the ‘launch’ button at the bottom.
Understanding Your Annotate Results¶
If all goes well, you should get the following:
- logs folder: This folder contains the ‘conder_stderr’ and ‘condor_stdout’ files. The files record feedback, progress and, importantly, any errors the app encountered during the analysis. You won’t normally need to look at these but they are very helpful in figuring out what may have happened if your output doesn’t look like you expected.
- seq_pep folder: This folder contains the BLAST database files used in your analysis.
- sqlite3 folder: This folder contains the annotation database files used in your analysis
- <species>.tsv: This is the tab-delimited output from the BLAST search. It is unlikely that you will need to look at this file.
- <output_prefix>.txt: KOBAS-annotate generates a text file with the name you provide. It has two sections.
- <output_prefix>.txt_KOBAS_acc_pathways.tsv: A tab-delimited file with accession number and all of the annoations made to that accession.
- <output_prefix>.txt_KOBAS_pathwyas_acc.tsv: A tab-delimited file with pathways and of all of the accession annotated with that pathway.
The first section of <output_prefix>.txt looks like this:
##dme Drosophila melanogaster (fruit fly)
##Method: BLAST Options: evalue <= 1e-05
##Summary: 87 succeed, 0 fail
#Query Gene ID|Gene name|Hyperlink
lcl|NW_020311285.1_prot_XP_012256083.1_15 dme:Dmel_CG34349|Unc-13-4B|http://www.genome.jp/dbget-bin/www_bget?dme:Dmel_CG34349
lcl|NW_020311286.1_prot_XP_020708336.1_46 dme:Dmel_CG6963|gish|http://www.genome.jp/dbget-bin/www_bget?dme:Dmel_CG6963
lcl|NW_020311285.1_prot_XP_020707987.1_39 dme:Dmel_CG30403||http://www.genome.jp/dbget-bin/www_bget?dme:Dmel_CG30403
The second section of <output_prefix>.txt follows a dashed line and looks like this:
--------------------
////
Query: lcl|NW_020311285.1_prot_XP_012256083.1_15
Gene: dme:Dmel_CG34349 Unc-13-4B
Entrez Gene ID: 43002
////
Query: lcl|NW_020311286.1_prot_XP_020708336.1_46
Gene: dme:Dmel_CG6963 gish
Entrez Gene ID: 49701
Pathway: Hedgehog signaling pathway - fly KEGG PATHWAY dme04341
////
Query: lcl|NW_020311285.1_prot_XP_020707987.1_39
Gene: dme:Dmel_CG30403
Entrez Gene ID: 246595
////
Query: lcl|NW_020311285.1_prot_XP_020707989.1_40
Gene: dme:Dmel_CG6148 Past1
Entrez Gene ID: 41569
Pathway: Endocytosis KEGG PATHWAY dme04144
Hemostasis Reactome R-DME-109582
Factors involved in megakaryocyte development and platelet production Reactome R-DME-98323
The <output_prefix>.txt_KOBAS_acc_pathways.tsv file looks like this:
XP_018223853.1 Reactome:R-SCE-6782135,KEGG:sce03420,Reactome:R-SCE-113418,Reactome:R-SCE-3700989,Reactome:R-SCE-73894,Reactome:R-SCE-73776,KEGG:sce03022,Reactome:R-SCE-6796648 XP_018222686.1 Reactome:R-SCE-5689603,KEGG:sce03050,Reactome:R-SCE-392499,Reactome:R-SCE-168249,Reactome:R-SCE-597592,Reactome:R-SCE-1236975,Reactome:R-SCE-1236978 XP_018223153.1 KEGG:sce01100,KEGG:sce01110,KEGG:sce01130,KEGG:sce01200,KEGG:sce01230,BioCyc:NONOXIPENT-PWY,BioCyc:PENTOSE-P-PWY,KEGG:sce00030 XP_018220571.1 KEGG:sce01100,KEGG:sce00270,KEGG:sce00480,KEGG:sce00410,KEGG:sce00330,BioCyc:ARGSPECAT-PWY
The <output_prefix>.txt_KOBAS_pathwyas_acc.tsv file looks like this:
KEGG:sce01100 XP_018223153.1,XP_018220571.1,XP_018219513.1 Reactome:R-SCE-5688426 XP_018222686.1 KEGG:sce03022 XP_018223853.1 Reactome:R-SCE-75105 XP_018219513.1 Reactome:R-SCE-597592 XP_018222686.1
If your analysis doesn’t complete as you expected please look at your ‘condor_stderr’ and ‘condor_stdout’ files. If that doesn’t clarify the problem contact us at agbase@email.arizona.edu or support@cyverse.org.
KOBAS identify 3.0.3¶
Launching the App¶
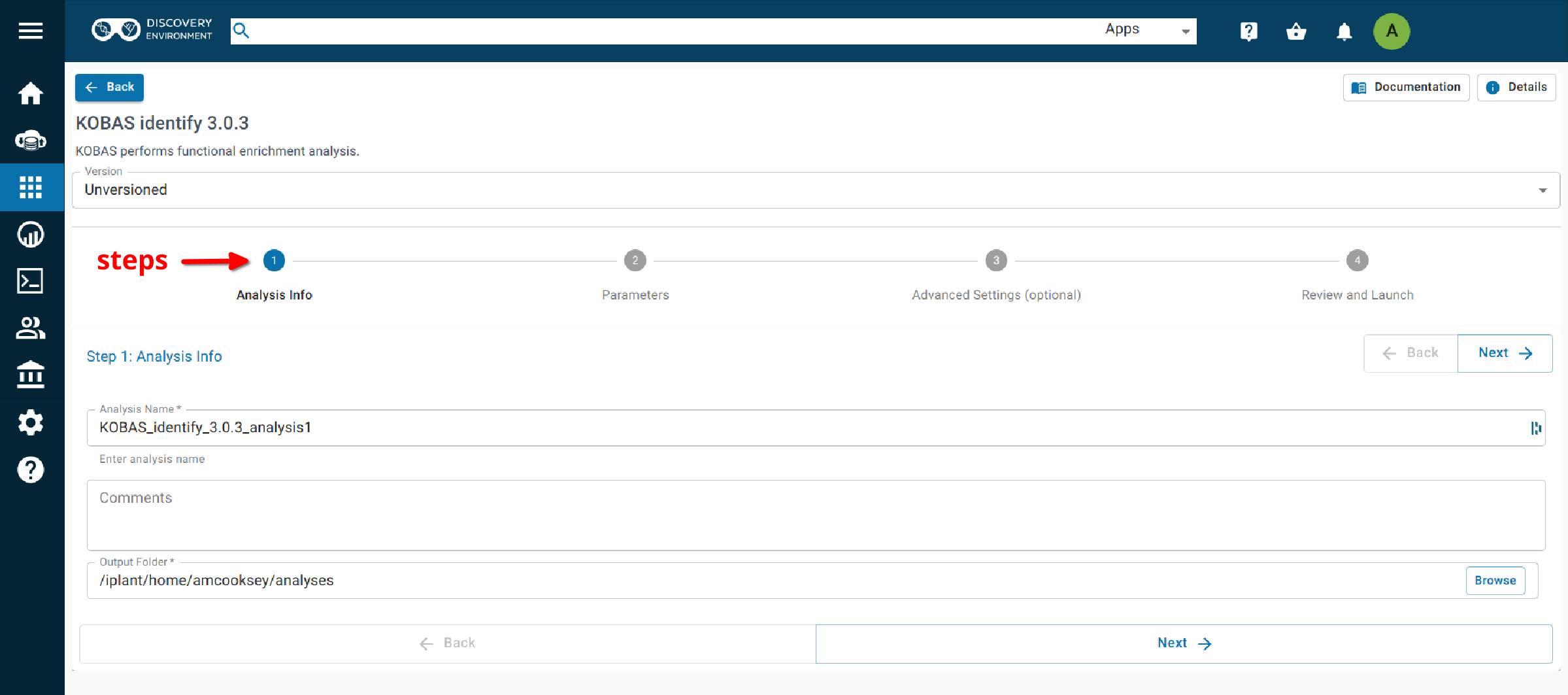
Step 1. Analysis Info¶
Analysis Name: KOBAS_identify_3.0.3_analysis_1: This menu is used to name the job you will run so that you can find it later. Analysis Name: The default name is “KOBAS_identify_3.0.3_analysis1”. We recommend changing the ‘analysis1’ portion of this to reflect the data you are running.
Comments: (Optional) You can add additional information in the comments section to distinguish your analyses further.
Select output folder: This is where your results will be placed. The default (recommended) is your ‘analyses’ folder.
Step 2. Parameters¶
Foreground File: Use the ‘browse’ button on the right side of the field to navigate to your input file. This should be the output of KOBAS annotate.
Background: Enter the species for the species of the sequences in your input file.
Note
If you don’t know the code for your species it can be found here: https://www.kegg.jp/kegg/catalog/org_list.html
If your species of interest is not available then you should choose the code for the closest-related species available
Cutoff: Annotation terms with less than cutoff number of genes are not used for statistical tests. Default is 5.
Method: Choose the statistical method to be used from the drop-down list. Default is hypergeometric/Fisher’s Exact.
FDR: Method for determining false discovery rate. Default is Benjamnini-Hochberg.
Output File Name: Provide an output file name.
Step3. Adavanced Settings (optional)¶
This page allows you specifiy compute requirements for your analysis (e.g. more memory if your analysis is particularly large). You should be able to leave the defaults for most analyses.
Step4. Review and Launch¶
This will display all of the parameters you have set (other than default). Missing information that is required will displayed in red. Make sure you are happy with your choices and then clicke the ‘launch’ button at the bottom.
Understanding Your Identify Results¶
If all goes well, you should get the following:
- logs folder: This folder contains the ‘conder_stderr’ and ‘condor_stdout’ files. The files record feedback, progress and, importantly, any errors the app encountered during the analysis. You won’t normally need to look at these but they are very helpful in figuring out what may have happened if your output doesn’t look like you expected.
- sqlite3 folder: This folder contains the annotation database files used in your analysis
- <output_file_name_you_provided>: KOBAS identify generates a text file with the name you provide.
##Databases: PANTHER, KEGG PATHWAY, Reactome, BioCyc
##Statistical test method: hypergeometric test / Fisher's exact test
##FDR correction method: Benjamini and Hochberg
#Term Database ID Input number Background number P-Value Corrected P-Value Input Hyperlink
Hedgehog signaling pathway - fly KEGG PATHWAY dme04341 12 33 3.20002656734e-18 1.76001461204e-16 lcl|NW_020311286.1_prot_XP_012256678.1_51|lcl|NW_020311286.1_prot_XP_025602973.1_48|lcl|NW_020311286.1_prot_XP_012256683.1_52|lcl|NW_020311286.1_prot_XP_012256679.1_55|lcl|NW_020311286.1_prot_XP_012256674.1_54|lcl|NW_020311286.1_prot_XP_020708336.1_46|lcl|NW_020311285.1_prot_XP_012256108.1_32|lcl|NW_020311286.1_prot_XP_012256682.1_53|lcl|NW_020311286.1_prot_XP_025603025.1_47|lcl|NW_020311286.1_prot_XP_020708334.1_49|lcl|NW_020311285.1_prot_XP_012256109.1_33|lcl|NW_020311286.1_prot_XP_020708333.1_50 http://www.genome.jp/kegg-bin/show_pathway?dme04341/dme:Dmel_CG6963%09red/dme:Dmel_CG6054%09red
Hedgehog signaling pathway PANTHER P00025 6 13 3.6166668094e-10 9.94583372585e-09 lcl|NW_020311286.1_prot_XP_025602279.1_78|lcl|NW_020311286.1_prot_XP_025602289.1_76|lcl|NW_020311286.1_prot_XP_025602264.1_79|lcl|NW_020311285.1_prot_XP_012256108.1_32|lcl|NW_020311285.1_prot_XP_012256109.1_33|lcl|NW_020311286.1_prot_XP_012256943.1_77 http://www.pantherdb.org/pathway/pathwayDiagram.jsp?catAccession=P00025
Signaling by NOTCH2 Reactome R-DME-1980145 3 8 2.00259649553e-05 0.000275357018136 lcl|NW_020311285.1_prot_XP_012256118.1_28|lcl|NW_020311285.1_prot_XP_012256117.1_27|lcl|NW_020311285.1_prot_XP_012256119.1_26 http://www.reactome.org/cgi-bin/eventbrowser_st_id?ST_ID=R-DME-1980145
If your analysis doesn’t complete as you expected please look at your ‘condor_stderr’ and ‘condor_stdout’ files. If that doesn’t clarify the problem contact us at agbase@email.arizona.edu or support@cyverse.org.
KOBAS annotate and identify 3.0.3¶
Launching the App¶
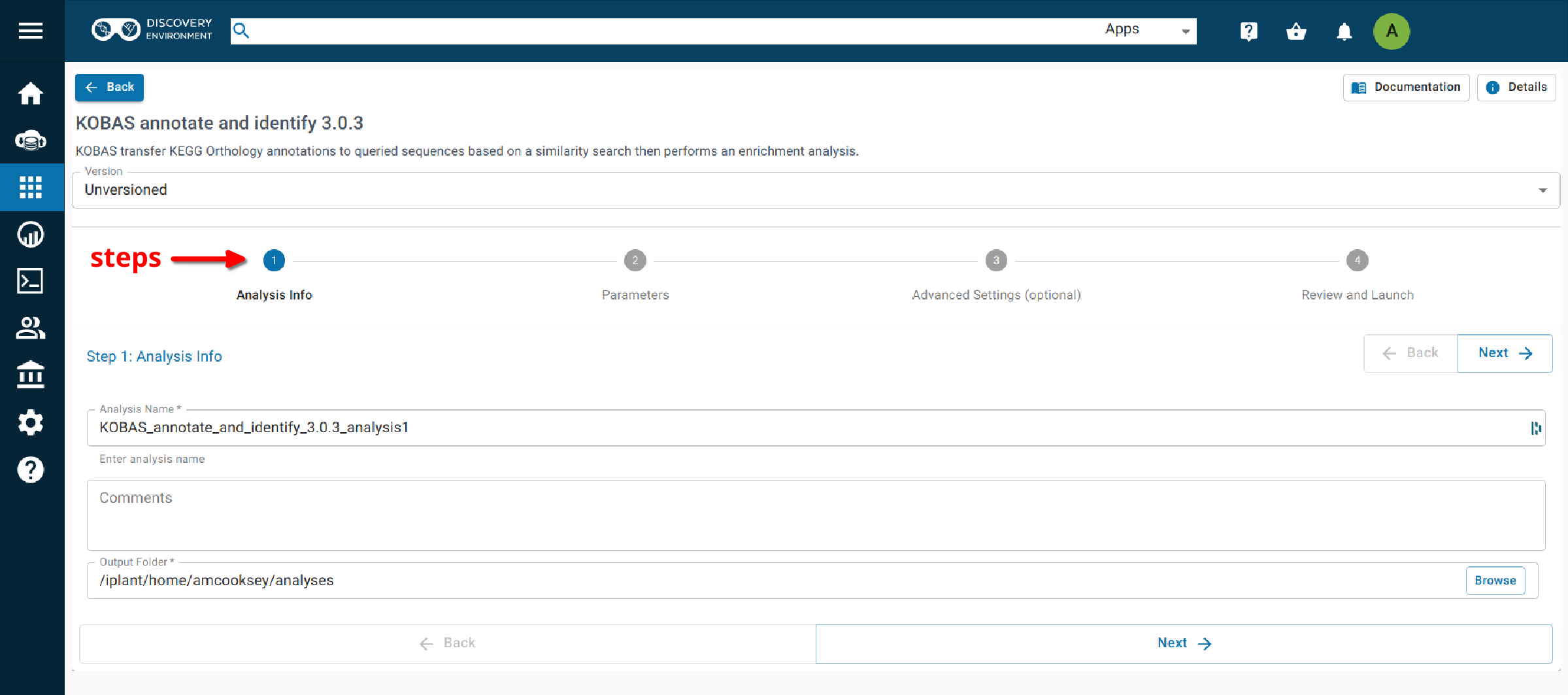
This app runs both the annotate and identify analyses together as a convenience for user who wish to run both steps.
Step 1. Analysis Info¶
Analysis Name: KOBAS_annotate_and_identify_3.0.3_analysis_1: This menu is used to name the job you will run so that you can find it later. Analysis Name: The default name is “KOBAS_annotate_identify_3.0.3_analysis1”. We recommend changing the ‘analysis1’ portion of this to reflect the data you are running.
Comments: (Optional) You can add additional information in the comments section to distinguish your analyses further.
Select output folder: This is where your results will be placed. The default (recommended) is your ‘analyses’ folder.
Step 2. Parameters¶
Input¶
Input File: Use the ‘browse’ button on the right side of the field to navigate to your input file.
Input File Type: Select your input file type from the drop-down list. If your file type isn’t there then the app does not support that file type.
Annotate Options¶
Species Code: Enter the species for the species of the sequences in your input file.
Note
If you don’t know the code for your species it can be found here: https://www.kegg.jp/kegg/catalog/org_list.html
If your species of interest is not available then you should choose the code for the closest-related species available
E value: This is the evalue to use in the BLAST search. The default is 1e-5.
Rank: rank cutoff for valid hits from BLAST result. Default is 5.
Covergage: subject coverage cutoff for BLAST. Default is 0.
Ortholog: when checked KOBAS will only use orthologs for cross species annotation.
Identify Options¶
Cutoff: Annotation terms with less than cutoff number of genes are not used for statistical tests. Default is 5.
Method: Choose the statistical method to be used from the drop-down list. Default is hypergeometric/Fisher’s Exact.
FDR: Method for determining false discovery rate. Default is Benjamnini-Hochberg.
Output¶
Output File Basename: This will the the prefix of your output files.
Step3. Adavanced Settings (optional)¶
This page allows you specifiy compute requirements for your analysis (e.g. more memory if your analysis is particularly large). You should be able to leave the defaults for most analyses.
Step4. Review and Launch¶
This will display all of the parameters you have set (other than default). Missing information that is required will displayed in red. Make sure you are happy with your choices and then clicke the ‘launch’ button at the bottom.
If your analysis doesn’t complete as you expected please look at your ‘condor_stderr’ and ‘condor_stdout’ files. If that doesn’t clarify the problem contact us at agbase@email.arizona.edu or support@cyverse.org.