InterProScan on CyVerse¶
Accessing InterProScan in the Discovery Environment¶
- Create an account on CyVerse (free)
- Open the CyVerse Discovery Environment (DE) and login with your CyVerse credentials.
- If you are new to the Discovery Environment (DE) the user guide can be found here.
- Click on the ‘Data’ button at the left side of the screen to access your files/folders. Upload your data to the DE.
- To access the InterProScan Sequence Search 5.36-75.0 app click on the ‘Apps’ button at the left side of the DE.
- Search for ‘interproscan’ in the search bar at the top of the ‘apps’ window. The contents of the folder will appear in the main pane of the window. The InterProScan app is called ‘InterProScan Sequence Search 5.36-75’; click on the name to open the app.
Using the InterProScan App¶
Launching the App¶
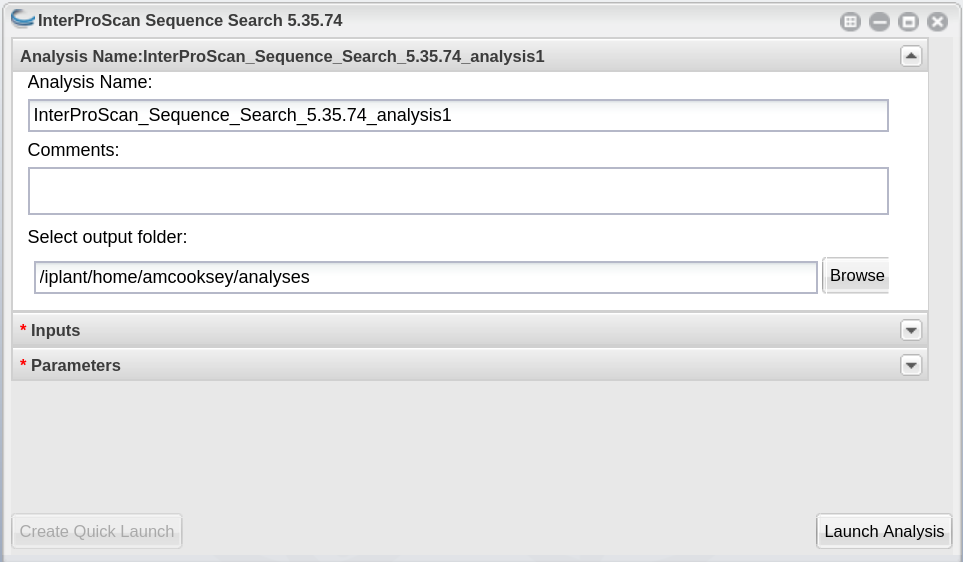
InterProScan_Sequence_Search_5.36.75_analysis1: This menu is used to name the job you will run so that you can find it later. Analysis Name: The default name is “InterProScan_Sequence_Search_5.36.75_analysis1”. We recommend changing the ‘analysis1’ portion of this to reflect the data you are running.
Comments: (Optional) You can add additional information in the comments section to distinguish your analyses further.
Select output folder: This is where your results will be placed. The default (recommended) is your ‘analyses’ folder.
Retain Inputs: Enabling this flag will copy all the input files into the analysis result folder.
Warning
Selecting this option will rapidly consume your allocated space. It is not recommended. Your inputs will always remain available in the folder in which you stored them.
Input¶
Peptide FASTA file: Use the Browse button on the right hand side to navigate to your Data folder and select your protein sequence file.
Parameters¶
Annotate each peptide with Gene Ontology information: Be sure this box is checked. This will ensure that you get GO annotations
Biocurator: This will be used to fill the ‘assigned by’ field of your GAF output file. If you do not fill it in the default “user” will be used instead.
Database: Use the database that sequences were obtained from (Genbank), or a recognizable project name if these sequences are not in a database (e.g., i5k project or Smith Lab).
Annotate each peptide with biological pathway information: This is optional. However, if you want pathways annotations be it is checked.
Taxon: Enter the NCBI taxon number for your species. This can be found by searching for your species name (common or scientific) in the NCBI taxon database.
InterProScan output directory name: This will be the name of the folder for your output files. The default folder name is ‘outdir’.
Understanding Your Results¶
InterProScan Outputs¶
This app provides all six of the InterProScan output formats. For more details on the contents of each file please refer to the InterProScan outputs documentation.
<basename>.gff3
<basename>.tsv
<basename>.xml
<basename>.json
<basename>.html.tar.gz
<basename>.svg.tar.gz
This app also runs the ‘InterProScan Results Function’ on the XML output from InterProScan. This tool provides a GAF output file and a variety of summary (count) files described below.
InterProScan Results Function Outputs¶
<basename>_gaf.txt: -This table follows the formatting of a gene association file (gaf) and can be used in GO enrichment analyses.
<basename>_acc_go_counts.txt: -This table includes input accessions, the number of GO IDs assigned to each accession and GO ID names. GO IDs are split into BP (Biological Process), MF (Molecular Function) and CC (Cellular Component).
<basename>_go_counts.txt: -This table counts the numbers of sequences assigned to each GO ID so that the user can quickly identify all genes assigned to a particular function.
<basename>_acc_interpro_counts.txt: -This table includes input accessions, number of InterPro IDs for each accession, InterPro IDs assigned to each sequence and the InterPro ID name.
<basename>_interpro_counts.txt: -This table counts the numbers of sequences assigned to each InterPro ID so that the user can quickly identify all genes with a particular motif.
<basename>_acc_pathway_counts.txt: -This table includes input accessions, number of pathway IDs for the accession and the pathway names. Multiple values are separated by a semi-colon.
<basename>_pathway_counts.txt: -This table counts the numbers of sequences assigned to each Pathway ID so that the user can quickly identify all genes assigned to a pathway.
<basename>.err: -This file will list any sequences that were not able to be analyzed by InterProScan. Examples of sequences that will cause an error are sequences with a large run of Xs.
If you output doesn’t look like you expect please check the ‘condor_stderr’ file in the analysis output ‘logs’ folder. If that doesn’t clarify the problem contact us at agbase@email.arizona.edu or support@cyverse.org.